

-
 EXPERTS INCosmetic injections. Skin Care.
EXPERTS INCosmetic injections. Skin Care.
Medical Lasers.
#1 Juvederm® Voluma in Georgia for 2015, 2016, & 2017
Restylane® Lyft 2018 -
 YOUNGER
YOUNGER
LOOKING
SKINIs only a visit away.Schedule a free consultation -
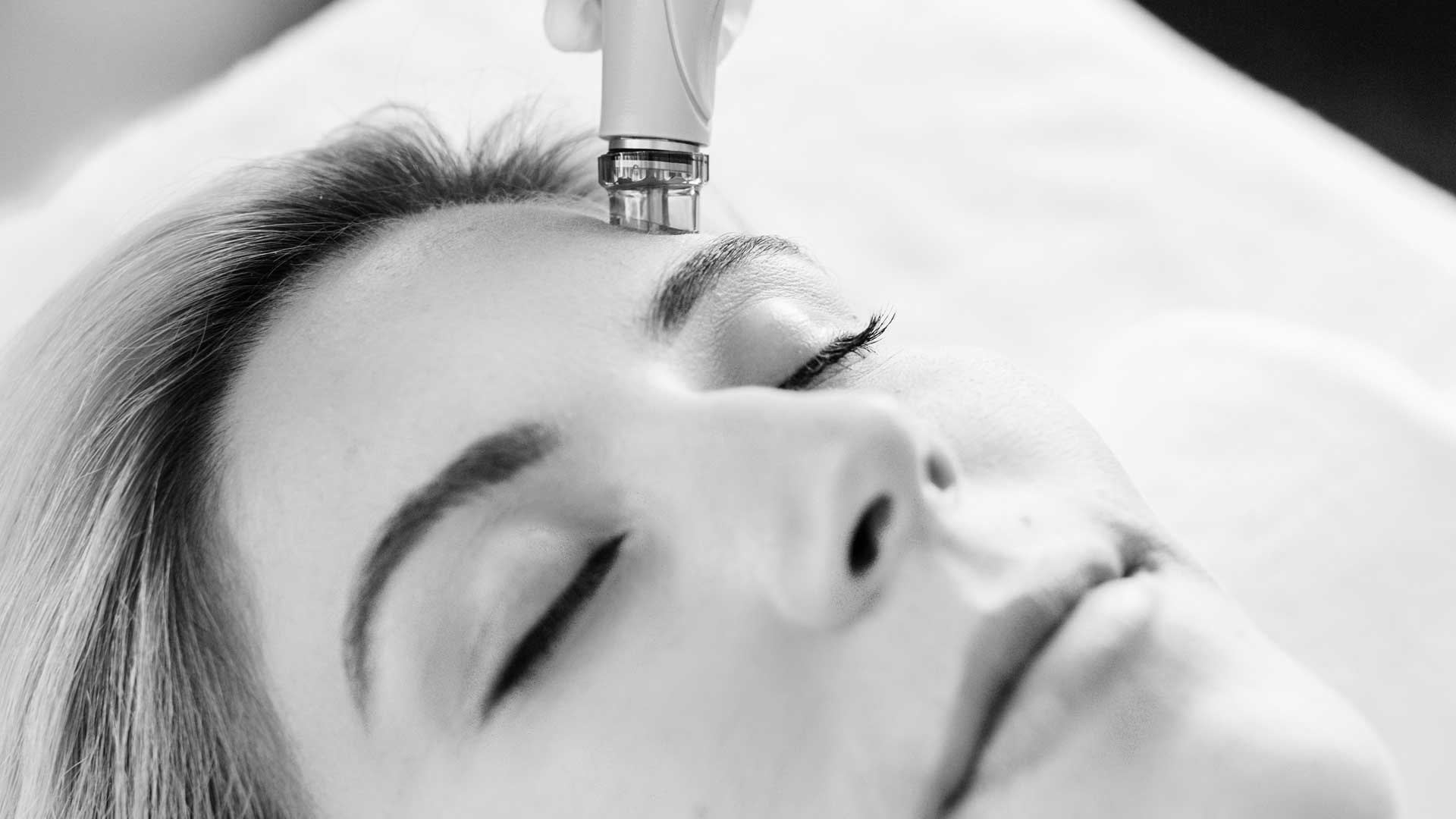 SKIN
SKIN
TREATMENTSHydrafacials, Chemical Peels, Microneedling, Microderm Abrasion, FacialsView More -
 INJECTABLES
INJECTABLES
#1 in GeorgiaBotox®, Juvederm®, Voluma®, and More!Learn More
Truffles Aesthetics is an Allergan® Top 50 provider. This means we are in the top 1% of all providers in the USA of Allergan® products such as of Botox®, JUVÉDERM®, and Kybella® products.
Injectables
Botox®, Voluma®, Juvéderm®, and more.
View more
Truffles offers top-selling, trusted, FDA approved injectables brands to our patients. Staff has been highly trained to use the products in a safe, effective manner to combat the most common early signs of aging.
Skin Treatment
Aging Skin, Wrinkles, Brown Spots, Rosacea, Acne
View more
Truffles is constantly adding new, innovative skin treatments to the portfolio of options we offer our patients. The skin of each patient is unique, so we customize each treatment based on the individual’s specific skincare needs.
Lasers
Hair Removal, Photofacials, Resurfacing and Tattoo Removal
View more
Truffles Aesthetics offers the latest in laser treatments. Lasers can be used to treat dark facial spots, facial veins, redness, remove tattoos and to remove unwanted hair.
Massages
Relieve Stress and Ease Aches.
View more
Take a break from the stress of modern life or work out those aches and pains from exercising or pregnancy with your choice of a variety of massages from our licensed massage therapist.
"Can't Wait to Go Back!"
Monthly Specials
Save Over 50%!
We offer a rotating list of amazing monthly specials to our clients on products and procedures they love. Specials include Botox®, Juvederm®, Voluma®, and More! Join our mailing list to enjoy our exclusive specials!
Indications
BOTOX® Cosmetic (onabotulinumtoxinA) is indicated in adult patients for the temporary improvement in the appearance of:
- moderate to severe glabellar lines associated with corrugator and/or procerus muscle activity
- moderate to severe lateral canthal lines associated with orbicularis oculi activity
- moderate to severe forehead lines associated with frontalis activity
IMPORTANT SAFETY INFORMATION, INCLUDING BOXED WARNING
WARNING: DISTANT SPREAD OF TOXIN EFFECT
Postmarketing reports indicate that the effects of BOTOX® Cosmetic and all botulinum toxin products may spread from the area of injection to produce symptoms consistent with botulinum toxin effects. These may include asthenia, generalized muscle weakness, diplopia, ptosis, dysphagia, dysphonia, dysarthria, urinary incontinence and breathing difficulties. These symptoms have been reported hours to weeks after injection. Swallowing and breathing difficulties can be life threatening and there have been reports of death. The risk of symptoms is probably greatest in children treated for spasticity but symptoms can also occur in adults treated for spasticity and other conditions, particularly in those patients who have an underlying condition that would predispose them to these symptoms. In unapproved uses, including spasticity in children, and in approved indications, cases of spread of effect have been reported at doses comparable to those used to treat cervical dystonia and spasticity and at lower doses.
CONTRAINDICATIONS
BOTOX® Cosmetic is contraindicated in the presence of infection at the proposed injection site(s) and in individuals with known hypersensitivity to any botulinum toxin preparation or to any of the components in the formulation.
WARNINGS AND PRECAUTIONS
Lack of Interchangeability between Botulinum Toxin Products
The potency Units of BOTOX® Cosmetic are specific to the preparation and assay method utilized. They are not interchangeable with other preparations of botulinum toxin products and, therefore, units of biological activity of BOTOX® Cosmetic cannot be compared to nor converted into units of any other botulinum toxin products assessed with any other specific assay method.
Spread of Toxin Effect
Please refer to Boxed Warning for Distant Spread of Toxin Effect.
No definitive serious adverse event reports of distant spread of toxin effect associated with dermatologic use of BOTOX® Cosmetic at the labeled dose of 20 Units (for glabellar lines), 24 Units (for lateral canthal lines), 40 Units (for forehead lines with glabellar lines), 44 Units (for simultaneous treatment of lateral canthal lines and glabellar lines), and 64 Units (for simultaneous treatment of lateral canthal lines, glabellar lines, and forehead lines) have been reported.
Serious Adverse Reactions With Unapproved Use
Serious adverse reactions, including excessive weakness, dysphagia, and aspiration pneumonia, with some adverse reactions associated with fatal outcomes, have been reported in patients who received BOTOX® injections for unapproved uses. In these cases, the adverse reactions were not necessarily related to distant spread of toxin, but may have resulted from the administration of BOTOX® to the site of injection and/or adjacent structures. In several of the cases, patients had pre-existing dysphagia or other significant disabilities. There is insufficient information to identify factors associated with an increased risk for adverse reactions associated with the unapproved uses of BOTOX®. The safety and effectiveness of BOTOX® for unapproved uses have not been established.
Hypersensitivity Reactions
Serious and/or immediate hypersensitivity reactions have been reported. These reactions include anaphylaxis, serum sickness, urticaria, soft-tissue edema, and dyspnea. If such reactions occur, further injection of BOTOX® Cosmetic should be discontinued and appropriate medical therapy immediately instituted. One fatal case of anaphylaxis has been reported in which lidocaine was used as the diluent and, consequently, the causal agent cannot be reliably determined.
Cardiovascular System
There have been reports following administration of BOTOX® of adverse events involving the cardiovascular system, including arrhythmia and myocardial infarction, some with fatal outcomes. Some of these patients had risk factors including pre-existing cardiovascular disease. Use caution when administering to patients with pre-existing cardiovascular disease.
Increased Risk of Clinically Significant Effects with Pre-existing Neuromuscular Disorders
Individuals with peripheral motor neuropathic diseases, amyotrophic lateral sclerosis, or neuromuscular junction disorders (eg, myasthenia gravis or Lambert-Eaton syndrome) should be monitored when given botulinum toxin. Patients with neuromuscular disorders may be at increased risk of clinically significant effects including generalized muscle weakness, diplopia, ptosis, dysphonia, dysarthria, severe dysphagia, and respiratory compromise from onabotulinumtoxinA (see Warnings and Precautions).
Dysphagia and Breathing Difficulties
Treatment with BOTOX® and other botulinum toxin products can result in swallowing or breathing difficulties. Patients with pre-existing swallowing or breathing difficulties may be more susceptible to these complications. In most cases, this is a consequence of weakening of muscles in the area of injection that are involved in breathing or oropharyngeal muscles that control swallowing or breathing (see Boxed Warning).
Pre-existing Conditions at the Injection Site
Caution should be used when BOTOX® Cosmetic treatment is used in the presence of inflammation at the proposed injection site(s) or when excessive weakness or atrophy is present in the target muscle(s).
Human Albumin and Transmission of Viral Diseases
This product contains albumin, a derivative of human blood. Based on effective donor screening and product manufacturing processes, it carries an extremely remote risk for transmission of viral diseases and variant Creutzfeldt-Jakob disease (vCJD). There is a theoretical risk for transmission of Creutzfeldt-Jakob disease (CJD), but if that risk actually exists, the risk of transmission would also be considered extremely remote. No cases of transmission of viral diseases, CJD or vCJD have ever been identified for licensed albumin or albumin contained in other licensed products.
ADVERSE REACTIONS
The most frequently reported adverse reaction following injection of BOTOX® Cosmetic for glabellar lines was eyelid ptosis (3%).
The most frequently reported adverse reaction following injection of BOTOX® Cosmetic for lateral canthal lines was eyelid edema (1%).
The most frequently reported adverse reactions following injection of BOTOX® Cosmetic for forehead lines with glabellar lines were headache (9%), brow ptosis (2%) and eyelid ptosis (2%).
DRUG INTERACTIONS
Co-administration of BOTOX® Cosmetic and aminoglycosides or other agents interfering with neuromuscular transmission (eg, curare-like compounds) should only be performed with caution as the effect of the toxin may be potentiated. Use of anticholinergic drugs after administration of BOTOX® Cosmetic may potentiate systemic anticholinergic effects.
The effect of administering different botulinum neurotoxin products at the same time or within several months of each other is unknown. Excessive neuromuscular weakness may be exacerbated by administration of another botulinum toxin prior to the resolution of the effects of a previously administered botulinum toxin.
Excessive weakness may also be exaggerated by administration of a muscle relaxant before or after administration of BOTOX® Cosmetic.
USE IN SPECIFIC POPULATIONS
There are no studies or adequate data from postmarketing surveillance on the developmental risk associated with use of BOTOX® Cosmetic in pregnant women. There are no data on the presence of BOTOX® Cosmetic in human or animal milk, the effects on the breastfed child, or the effects on milk production.
Please see BOTOX® Cosmetic full BOTOXcosmetic" target="_blank">Prescribing Information including Boxed Warning and Medication Guide.
CoolSculpting® Treatment Important Information
Indications
The CoolSculpting® procedure is FDA-cleared for the treatment of visible fat bulges in the thigh, abdomen and flank, along with bra fat, back fat, underneath the buttocks (also known as banana roll), and upper arm in patients with a Body Mass Index (BMI) of ≤ 30 and in submental and submandibular areas in patients with a BMI of ≤ 46.2. It is also FDA-cleared to affect the appearance of lax tissue with submental area treatments.
Important Safety Information
CoolSculpting® is contraindicated in patients with cryoglobulinemia, cold agglutinin disease, or paroxysmal cold hemoglobinuria.
Ask your patient about any medical conditions including recent surgery, pre-existing hernia, and any known sensitivities or allergies.
During the procedure patients may experience sensations of pulling, tugging, mild pinching, intense cold, tingling, stinging, aching, and cramping at the treatment site. These sensations subside as the area becomes numb. Following the procedure, typical side effects include temporary redness, swelling, blanching, bruising, firmness, tingling, stinging, tenderness, cramping, aching, itching, or skin sensitivity, and sensation of fullness in the back of the throat after submental or submandibular area treatment.
Rare side effects may also occur. Paradoxical hyperplasia (visibly enlarged tissue volume in the treated area) may develop 2-5 months after treatment and requires surgical intervention for correction.
As with any medical procedure, a consultation should be done by a licensed healthcare professional to determine if the patient is a candidate for treatment. Consult the CoolSculpting® System User Manual for a complete list of Contraindications, Warnings, Precautions, and potential side effects. Treatment applications that deviate from the guidelines are not recommended.
JUVÉDERM® Collection of Fillers Important Information
INDICATIONS
JUVÉDERM VOLUMA® XC injectable gel is indicated for deep (subcutaneous and/or supraperiosteal) injection for cheek augmentation to correct age-related volume deficit in the mid-face in adults over the age of 21.
JUVÉDERM® Ultra XC and JUVÉDERM® Ultra Plus XC injectable gels are indicated for injection into the mid-to-deep dermis for correction of moderate to severe facial wrinkles and folds (such as nasolabial folds).
JUVÉDERM VOLLURE™ XC injectable gel is indicated for injection into the mid-to-deep dermis for correction of moderate to severe facial wrinkles and folds (such as nasolabial folds) in adults over the age of 21.
JUVÉDERM® Ultra XC injectable gel is indicated for injection into the lips and perioral area for lip augmentation in adults over the age of 21.
JUVÉDERM VOLBELLA® XC injectable gel is indicated for injection into the lips for lip augmentation and for correction of perioral rhytids in adults over the age of 21.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
These products should not be used in patients who have severe allergies, marked by a history of anaphylaxis or history or presence of multiple severe allergies, and should not be used in patients with a history of allergies to Gram-positive bacterial proteins or lidocaine contained in these products.
- Do not inject into blood vessels. Introduction of these products into the vasculature may lead to embolization, occlusion of the vessels, ischemia, or infarction. Take extra care when injecting soft-tissue fillers; for example, inject the product slowly and apply the least amount of pressure necessary. Rare, but serious, adverse events associated with the intravascular injection of soft-tissue fillers in the face have been reported and include temporary or permanent vision impairment, blindness, cerebral ischemia or cerebral hemorrhage leading to stroke, skin necrosis, and damage to underlying facial structures. Immediately stop the injection if a patient exhibits any of the following symptoms: changes in vision, signs of a stroke, blanching of the skin, unusual pain during or shortly after the procedure. Patients should receive prompt medical attention and, possibly, evaluation by an appropriate healthcare professional specialist should an intravascular injection occur
- Product use at specific sites in which an active inflammatory process (skin eruptions such as cysts, pimples, rashes, or hives) or infection is present should be deferred until the underlying process has been controlled
- In order to minimize the risk of potential complications, these products should only be used by healthcare professionals who have appropriate training, experience, and knowledge of facial anatomy
- Healthcare professionals are encouraged to discuss the potential risks of soft-tissue injections with their patients prior to treatment and ensure that patients are aware of signs and symptoms of potential complications
- The safety and effectiveness for the treatment of anatomic regions other than the mid-face with JUVÉDERM VOLUMA® XC; facial wrinkles and folds with JUVÉDERM® Ultra XC, JUVÉDERM® Ultra Plus XC, and JUVÉDERM VOLLURE™ XC; and the lips and perioral area with JUVÉDERM® Ultra XC and JUVÉDERM VOLBELLA® XC have not been established in controlled clinical studies
- As with all transcutaneous procedures, dermal filler implantation carries a risk of infection. Follow standard precautions associated with injectable materials
- The safety for use during pregnancy, in breastfeeding females, and in patients with known susceptibility to keloid formation, hypertrophic scarring, and pigmentation disorders has not been studied
- The safety for use of JUVÉDERM VOLUMA® XC in patients under 35 or over 65 years, JUVÉDERM® Ultra XC and JUVÉDERM® Ultra Plus XC in patients under 18 years, and JUVÉDERM VOLLURE™ XC and JUVÉDERM VOLBELLA® XC in patients under 22 years has not been established
- Use with caution in patients on immunosuppressive therapy
- Patients who are using products that can prolong bleeding (such as aspirin, nonsteroidal anti-inflammatory drugs, and warfarin) may experience increased bruising or bleeding at treatment sites
- If laser treatment, chemical peel, or any other procedure based on active dermal response is considered after treatment, or if these products are administered before the skin has healed completely, there is a possible risk of an inflammatory reaction at the treatment site
- Patients who experience skin injury near the site of implantation may be at a higher risk for adverse events
- The safety of JUVÉDERM VOLUMA® XC injectable gel for use in patients with very thin skin in the mid-face has not been established
- Patients may experience late onset nodules with use of dermal fillers, including JUVÉDERM VOLUMA® XC
- Patients may experience late onset adverse events with use of dermal fillers
ADVERSE EVENTS
The most commonly reported side effects for JUVÉDERM® injectable gels were injection-site redness, swelling, pain, tenderness, firmness, lumps/bumps, bruising, discoloration, and itching. For JUVÉDERM VOLBELLA® XC, dryness was also reported. For JUVÉDERM VOLUMA® XC, side effects were predominantly moderate in severity, with duration of 2 to 4 weeks; for JUVÉDERM® Ultra XC, JUVÉDERM® Ultra Plus XC, or JUVÉDERM VOLLURE™ XC, they were mostly mild or moderate in severity, with duration of 14 days or less; and for JUVÉDERM VOLBELLA® XC, they were predominantly mild or moderate, with duration of 30 days or less.
To report an adverse reaction with any product in the JUVÉDERM® Collection, please call Allergan at 1-800-433-8871. Please visit JuvedermDFU.com for more information.
Products in the JUVÉDERM® Collection are available by prescription only.
KYBELLA® (deoxycholic acid) injection 10 mg/mL Important Information
INDICATION
KYBELLA® (deoxycholic acid) injection is indicated for improvement in the appearance of moderate to severe convexity or fullness associated with submental fat in adults.
The safe and effective use of KYBELLA® for the treatment of subcutaneous fat outside the submental region has not been established and is not recommended.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
KYBELLA® is contraindicated in the presence of infection at the injection sites.
WARNINGS AND PRECAUTIONS
Marginal Mandibular Nerve Injury
Cases of marginal mandibular nerve injury, manifested as an asymmetric smile or facial muscle weakness, were reported in 4% of subjects in the clinical trials; all cases resolved spontaneously (range 1-298 days, median 44 days). KYBELLA® should not be injected into or in close proximity to the marginal mandibular branch of the facial nerve.
Dysphagia
Dysphagia occurred in 2% of subjects in the clinical trials in the setting of administration-site reactions, eg, pain, swelling, and induration of the submental area; all cases of dysphagia resolved spontaneously (range 1-81 days, median 3 days). Avoid use of KYBELLA® in patients with current or prior history of dysphagia as treatment may exacerbate the condition.
Injection-Site Hematoma/Bruising
In clinical trials, 72% of subjects treated with KYBELLA® experienced hematoma/bruising. KYBELLA® should be used with caution in patients with bleeding abnormalities or who are currently being treated with antiplatelet or anticoagulant therapy as excessive bleeding or bruising in the treatment area may occur.
Risk of Injecting Into or in Proximity to Vulnerable Anatomic Structures
To avoid the potential of tissue damage, KYBELLA® should not be injected into or in close proximity (1 cm-1.5 cm) to salivary glands, lymph nodes, and muscles.
Injection Site Alopecia
Cases of injection site alopecia have been reported with administration of KYBELLA®. Onset and duration may vary among individuals and may persist. Consider withholding subsequent treatments until resolution.
Injection Site Ulceration and Necrosis
Injections that are too superficial into the dermis may result in skin ulceration and necrosis. Cases of injection site ulceration and necrosis have been reported with administration of KYBELLA®. Do not administer KYBELLA® into affected area until complete resolution.
ADVERSE REACTIONS
The most commonly reported adverse reactions in the pivotal clinical trials were: injection site edema/swelling, hematoma/bruising, pain, numbness, erythema, and induration.
Please see KYBELLA® full Prescribing Information.
LATISSE® (bimatoprost ophthalmic solution) 0.03% Important Information
Indication
LATISSE® (bimatoprost ophthalmic solution) 0.03% is indicated to treat hypotrichosis of the eyelashes by increasing their growth, including length, thickness, and darkness.
Important Safety Information
Contraindications: LATISSE® is contraindicated in patients with hypersensitivity to bimatoprost or to any of the ingredients.
Warnings and Precautions: In patients using LUMIGAN® (bimatoprost ophthalmic solution) or other prostaglandin analogs for the treatment of elevated intraocular pressure (IOP), the concomitant use of LATISSE® may interfere with the desired reduction in IOP. Patients using prostaglandin analogs including LUMIGAN® for IOP reduction should only use LATISSE® after consulting with their physician and should be monitored for changes to their intraocular pressure.
Increased iris pigmentation has occurred when bimatoprost solution was administered. Patients should be advised about the potential for increased brown iris pigmentation, which is likely to be permanent.
Bimatoprost has been reported to cause pigment changes (darkening) to periorbital pigmented tissues and eyelashes. The pigmentation is expected to increase as long as bimatoprost is administered, but has been reported to be reversible upon discontinuation of bimatoprost in most patients.
There is the potential for hair growth to occur in areas where LATISSE® solution comes in repeated contact with skin surfaces. Apply LATISSE® only to the skin of the upper eyelid margin at the base of the eyelashes.
LATISSE® solution should be used with caution in patients with active intraocular inflammation (eg, uveitis) because the inflammation may be exacerbated. LATISSE® should be used with caution in aphakic patients, in pseudophakic patients with a torn posterior lens capsule, or in patients with known risk factors for macular edema.
Adverse Reactions: The most frequently reported adverse events were eye pruritus, conjunctival hyperemia, skin hyperpigmentation, ocular irritation, dry eye symptoms, and periorbital erythema. These reactions occurred in less than 4% of patients.
Postmarketing Experience: The following adverse reactions have been identified during postapproval use of LATISSE®: dry skin of the eyelid and/or periocular area, eye swelling, eyelid edema, hypersensitivity (local allergic reactions), lacrimation increased, madarosis and trichorrhexis (temporary loss of a few eyelashes to loss of sections of eyelashes, and temporary eyelash breakage, respectively), periorbital and lid changes associated with a deepening of the eyelid sulcus, rash (including macular and erythematous), skin discoloration (periorbital), and vision blurred.
Please see LATISSE® full Prescribing Information.
REVOLVE™ Advanced Adipose System
Indications and Important Safety Information
INDICATIONS
The REVOLVE™ Advanced Adipose System (REVOLVE™ System) is used for aspiration, harvesting, filtering, and transferring of autologous adipose tissue for aesthetic body contouring. This system should be used with a legally marketed vacuum or aspirator apparatus as a source of suction. If harvested fat is to be re-implanted, the harvested fat is only to be used without any additional manipulation. REVOLVE™ System is intended for use in the following surgical specialties when the aspiration of soft tissue is desired: plastic and reconstructive surgery, gastrointestinal and affiliated organ surgery, urological surgery, general surgery, orthopedic surgery, gynecological surgery, thoracic surgery, and laparoscopic surgery.
IMPORTANT SAFETY INFORMATION
CONTRAINDICATIONS
Contraindications to autologous fat transfer include the presence of any disease processes that adversely affect wound healing, and poor overall health status of the individual.
WARNINGS
REVOLVE™ System must be used within the same surgical procedure. Reuse of this device in the same patient in a subsequent surgical procedure, or for more than one patient, may result in infection and/or transmission of communicable diseases. Do not use the product if sterile packaging is damaged.
This device will not, in and of itself, produce significant weight reduction. This device should be used with extreme caution in patients with chronic medical conditions such as diabetes, heart, lung, or circulatory system disease or obesity. The volume of blood loss and endogenous body fluid loss may adversely affect intra and/or postoperative hemodynamic stability and patient safety. The capability of providing adequate, timely replacement is essential for patient safety.
PRECAUTIONS
REVOLVE™ System is designed to remove localized deposits of excess fat through small incision and subsequently transfer the tissue back to the patient. Use of this device is limited to those physicians who, by means of formal professional training or sanctioned continuing medical education (including supervised operative experience), have attained proficiency in suction lipoplasty and tissue transfer. Results of this procedure will vary depending upon patient age, surgical site, and experience of the physician. Results of this procedure may or may not be permanent. The amount of fat removed should be limited to that necessary to achieve a desired cosmetic effect. Filling the device with adipose tissue over the maximum fill volume line can lead to occlusion of the mesh resulting in mesh tear.
ADVERSE EFFECTS
Some common adverse effects associated with autologous fat transfer are asymmetry, over- and/or under-correction of the treatment site, tissue lumps, bleeding, and scarring. Potential adverse effects associated with REVOLVE™ System include fat necrosis, cyst formation, infection, chronic foreign body response, allergic reaction, and inflammation.
REVOLVE™ System is available by prescription only.
For more information, please see the Instructions for Use (IFU) and User Manual for REVOLVE™ System.
To report an adverse reaction, please call Allergan at 1.800.367.5737.